
Você sabe o que é síndrome de Marfan? Conhecida também por aracnodactilia, a síndrome de Marfan é uma doença genética (ver também – Cardiomiopatia Hipertrófica: Sintomas, diagnóstico e tratamento) que acomete o tecido conjuntivo. Ela foi descrita pela primeira vez em 1896. Nesse post você conhecerá todos os detalhes desta doença, desde sinais/sintomas, diagnóstico e tratamento.
O que é a síndrome de Marfan?
A síndrome de Marfan é um distúrbio hereditário que afeta o tecido conjuntivo . Mas você sabe a função do tecido conjuntivo? Sua função é fazer a conexão entre os tecidos, fornecendo força e flexibilidade às estruturas como vasos sanguíneos, válvulas cardíacas, músculos e ossos.
A síndrome de Marfan afeta mais comumente o coração, os olhos, os vasos sanguíneos e o ossos. As pessoas com esta síndrome são geralmente altas e magras, com braços, pernas, dedos das mãos e pés incomumente longos.
O que causa a síndrome de Marfan?
A síndrome de Marfan é uma doença genética. Ela é causada por um defeito no gene ( FBN1) que permite que seu corpo produza uma proteína (fibrilina) que ajuda a dar elasticidade e força ao tecido conjuntivo. As alterações do FBN1 podem fazer com que os ossos cresçam demais, resultando em membros longos e altura significativa. Mas nem todos com essa alteração genética desenvolverão a síndrome de Marfan.
A maioria das pessoas herda o gene anormal de um pai que tem o distúrbio. Em cerca de 25% das pessoas com síndrome de Marfan, o gene anormal não vem de nenhum dos pais. Nesses casos, o gene sofre mutação pela primeira vez no óvulo ou no esperma dos pais. Esse gene mutante pode ser passado para a criança, que então desenvolverá a síndrome.
Existe alguma fator de risco para a Síndrome de Marfan?
A síndrome de Marfan afeta igualmente homens e mulheres e ocorre em todas as raças e grupos étnicos. Por ser uma condição genética, o maior fator de risco para a síndrome de Marfan é ter um dos pais com o distúrbio.
Quais são os sintomas da Síndrome de Marfan?
Os sinais e sintomas da síndrome de Marfan podem variar muito, mesmo entre membros da mesma família. Isto ocorre porque esta síndrome pode afetar muitas áreas diferentes do corpo. Algumas pessoas experimentam apenas efeitos leves, mas outras desenvolvem complicações com risco de vida.
Dentre as características mais importantes destacamos:
- Estatura alta com braços, pernas e dedos desproporcionalmente longos
- Alterações na caixa torácica como peito escavado ou que se projeta para fora
- Desvios na coluna como escoliose e cifose;
- Pé chato
- Um palato alto e arqueado e dentes apinhados
- Dilatação da raiz da aorta com possibilidade de aneurisma ( ruptura da aorta)
- Miopia extrema
- Subluxação do cristalino
Problemas oculares
As complicações oculares podem incluir:
Miopia e Astigmatismo – A maioria das pessoas com síndrome de Marfan apresentam miopia e astigmatismo. Estes podem ser notavelmente altos, pois o defeito do tecido conjuntivo pode afetar a córnea, o cristalino e o crescimento do olho
Subluxação do cristalino – O termo médico para esse problema é ectopia lentis, e significa uma mudança na localização da lente (cristalino) dentro do olho, chamada subluxação. Ectopia lentis ocorre em aproximadamente 60% dos indivíduos com síndrome de Marfan e é um dos principais critérios para o diagnóstico clínico dessa condição.
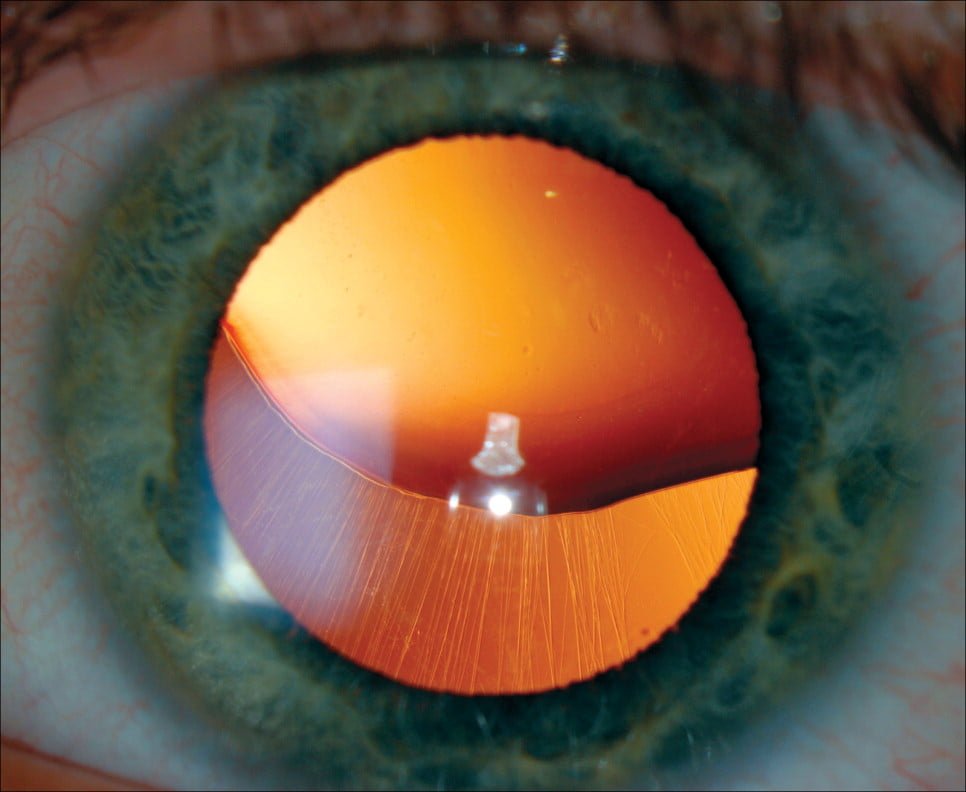
Glaucoma ou catarata de início precoce – As pessoas que têm a síndrome de Marfan tendem a desenvolver esses problemas oculares em uma idade mais jovem. O glaucoma faz com que a pressão dentro do olho aumente, o que pode danificar o nervo óptico. Cataratas são áreas turvas na lente normalmente clara do olho.
Descolamento da retina – existe o aumento do risco de descolamento ou ruptura na retina, o tecido sensível à luz que reveste a parede posterior do olho
Problemas musculoesqueléticos
As pessoas com síndrome de Marfan são mais altas que o esperado para a sua idade. Além disso, a amplitude dos braços (a distância entre as pontas dos dedos com os braços estendidos) é superior à altura delas.
Outra característica frequente é a aracnodactilia (dedos finos e desproporcionalmente longos). Ela é perceptível, com frequência, pelo sinal do polegar ou sinal do punho (imagem abaixo). Além do mais, é comum a deformidade do osso esterno — pectus carinatum (projeção para fora) ou pectus excavatum (projeção para dentro)
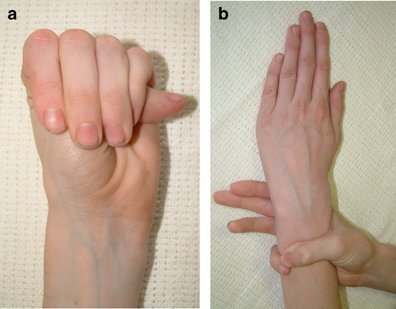
A síndrome de Marfan aumenta o risco de curvas anormais na coluna, como escoliose. Também pode interferir no desenvolvimento normal das costelas, o que pode fazer com que o esterno se projete ou pareça afundado no peito.
Problemas cardíacos e dos vasos sanguíneos
Cerca de 90% das pessoas com síndrome de Marfan desenvolvem alterações no coração e/ou nos vasos sanguíneos. E as complicações mais perigosas da síndrome de Marfan envolvem o aspecto cardiovascular.
As alterações que podem se desenvolver incluem:
Aneurisma da aorta. As paredes da aorta, a principal artéria que transporta o sangue do coração para o resto do corpo, tornam-se fracas, protuberantes e podem romper (explodir). Em pessoas com síndrome de Marfan, é mais provável que isso aconteça na raiz da aorta – onde a artéria sai do coração.
Dissecção aórtica. A parede da aorta é composta de camadas. Esta é uma ruptura na camada interna das três camadas da parede da aorta. O rasgo permite que o sangue entre na camada intermediária, o que estende o rasgo e leva a uma separação adicional e possivelmente à ruptura da parede. . Isso pode causar dor intensa no peito ou nas costas
Problemas nas válvulas cardíacas. A síndrome de Marfan pode fazer com que o tecido da válvula se torne fraco e estique. Consequentemente, isso leva ao mal funcionamento das válvulas Quando as válvulas cardíacas não funcionam adequadamente, seu coração geralmente precisa trabalhar mais para compensar. Isso pode eventualmente levar à insuficiência cardíaca.
Ritmo cardíaco anormal. A arritmia pode ocorrer em algumas pessoas com síndrome de Marfan. Muitas vezes está relacionado ao prolapso da válvula mitral.
Problemas bucais e dentários
As principais alterações bucais descritas incluem:
- Hipoplasia maxilar
- Dentes altamente apinhados com mordidas cruzadas
- Palato alto e profundo
Problemas pulmonares
As alterações no tecido pulmonar que ocorrem com a síndrome de Marfan aumentam o risco de:
- Asma.
- Doença pulmonar obstrutiva crônica (DPOC).
- Pneumonia.
- Pulmão colapsado (pneumotórax).
Como é feito o diagnóstico da Síndrome de Marfan?
Na maioria dos casos, o diagnóstico será baseado em um exame físico completo e uma avaliação detalhada do histórico médico e familiar.
A síndrome pode ser um desafio para os médicos diagnosticarem porque muitos distúrbios do tecido conjuntivo apresentam sinais e sintomas semelhantes. Mesmo entre membros da mesma família, os sinais e sintomas da síndrome de Marfan variam amplamente – tanto em suas características quanto em sua gravidade.
Logo, além do histórico detalhado, exames cardiológicos, oftalmológicos e genéticos são importantes para a confirmação do diagnóstico.
Quais exames são realizados?
Testes cardíacos
Se o seu médico suspeitar da síndrome de Marfan, um dos primeiros exames que ele pode recomendar é um ecocardiograma. Este exame utiliza as ondas sonoras para produzir uma imagem do seu coração, permitindo que os médicos verifiquem problemas no coração e na artéria aorta. Outras opções de imagem do coração incluem tomografia computadorizada (TC) e ressonância magnética (MRI).
Testes oculares
Os exames oftalmológicos são realizados por um médico especializado em doenças oculares (oftalmologista) para verificar o deslocamento da lente e a pressão do olho. Dentre os exames realizados destacam-se:
Exame de lâmpada de fenda. Este teste verifica deslocamento da lente, catarata ou retina descolada.
Teste de pressão ocular. Esse exame é para verificar se há glaucoma – aumento da pressão ocular
Teste genético
Utiliza-se o teste genético é frequentemente para confirmar o diagnóstico da síndrome de Marfan. É capaz de detectar um erro que causa a síndrome em 99% dos afetados.. Além disso, se uma mutação de Marfan for encontrada, os membros da família podem ser testados para ver se também são portadores da mutação. No entanto, além do exame ser caro, ele leva 3 meses para ser concluído.
Como é o tratamento da síndrome de Marfan?
Embora não haja cura para a síndrome de Marfan, a abordagem depende de quais partes do corpo são afetadas e da gravidade de sua condição.
Medicamentos
Medicamentos não são usados para tratar a síndrome de Marfan, mas podem ser usados para prevenir ou controlar complicações. Os medicamentos podem incluir:
Beta-bloqueadores – podem ajudar a tratar sintomas cardiovasculares. Eles podem diminuir a pressão no sistema cardiovascular, reduzindo a força e a frequência das contrações cardíacas. A terapia com betabloqueadores deve começar em uma idade precoce.
Bloqueadores dos receptores da angiotensina: Os bloqueadores dos receptores da angiotensina (BRA) são usados para tratar pressão alta e insuficiência cardíaca. Ensaios clínicos recentes mostraram que os BRAs ajudam a retardar o alargamento da aorta, assim como os betabloqueadores.
Tratamento para correção da aorta.
As alterações do tecido conjuntivo apresentam risco aumentado de complicações na aorta torácica (dissecção ou ruptura). Logo, a correção cirúrgica do aneurisma de aorta é fundamental em alguns casos. Seguem as indicações de cirurgia:
- Diâmetro da aorta ascendente > 50 mm
- Velocidade de crescimento transversal do aneurisma maior que 0,5cm/ano
- História familiar de dissecção de aorta com diâmetro de aorta < 50 mm
- Presença de insuficiência aórtica grave
Tratamento de escoliose
Quando há escoliose significativa, é necessária uma consulta com um especialista em coluna. Órtese e cirurgia são necessários em alguns casos. Além disso, existem opções cirúrgicas disponíveis para corrigir a aparência de um esterno afundado ou saliente.
Tratamento oftalmológico
Se você tem catarata, seu cristalino opaco pode ser substituído por um cristalino artificial. Além disso, em caso de deslocamento do cristalino ou descolamento da retina, o reparo cirúrgico é o tratamento de escolha.
Síndrome de Marfan e expectativa de vida
Devido aos avanços médicos (especialmente cirurgias cardíacas), algumas pessoas com síndrome de Marfan podem viver além dos 72 anos. O diagnóstico em uma idade precoce é fundamental porque o seu médico pode já iniciar o tratamento correto e retardar as complicações da doença.
Quem tem Síndrome de Marfan pode ter filhos?
O aconselhamento genético antes da gravidez é muito importante porque a síndrome de Marfan é uma condição hereditária. Pessoas grávidas e com síndrome de Marfan são consideradas casos de alto risco.
Se a sua aorta for de tamanho normal, o risco de dissecção é menor, mas ainda existe. Aqueles com aumento mesmo leve correm maior risco e o estresse da gravidez pode causar dilatação mais rapidamente. Por isso, durante a gravidez, você precisará de um acompanhamento cuidadoso com verificações frequentes da pressão arterial e ecocardiogramas mensais.
Quem tem síndrome de Marfan pode fazer academia?
As diretrizes de atividade física variam conforme os sintomas e a extensão da doença. A maioria das pessoas com síndrome de Marfan pode participar de algum tipo de atividade física e/ou recreativa. Mas se você tem uma aorta aumentada, provavelmente precisará evitar esportes de alta intensidade ou esportes de contato e exercícios isométricos (como levantamento de peso).
Cardiologista – Curitiba
A Loyola e Avellar possui profissionais capacitados e tem como objetivo cuidar da saúde e bem-estar de seus pacientes. Agende sua consulta agora mesmo: 41.3076-3054 ou whatsapp